

Mucopolysacharidóza je celá řada geneticky určených (podmíněných) patologií, které se objevují v případě poškození metabolismu kyselých mukopolysacharidů (glykosaminoglykanů).
Nejčastěji s takovým porušením je kostra postižena a fyzický vývoj je zpožděn a některé typy patologie se projevují zaostalostí intelektuální sféry. Kromě toho dochází k poruchám v kardiovaskulárním, gastrointestinálním a nervovém systému, v optickém aparátu a kůži.
Léčba mukopolysacharidózy je symptomatická, prognóza je obecně nepříznivá - narušuje se progrese, protože se akumulují glukosaminoglykany. Pacienti zemřou dost brzy.
Obecné údaje
Mucopolysacharidóza byla studována poměrně nedávno - v 20. století. Zpočátku to bylo považováno za jednu patologii, a proto kvůli různorodosti začaly rozlišovat své druhy. Sjednocujícím faktorem všech mukopolysacharidóz je to, že kyselinové mukopolysacharidy se hromadí v orgánech a tkáních a příčinou tohoto porušení je podřadnost některých buněčných enzymů, která je začleněna do genotypu a je zděděna.
Dávejte pozorOrtopedové léčí pacienty s mukopolysacharidózou, ale také vyžaduje klinické úsilí od kardiologů, oftalmologů, neurologů, otolaryngologů a některých dalších odborníků..
Mukopolysacharidóza typu IH
Mucopolysacharidóza typu IH se také nazývá Hurlerův syndrom. On je diagnostikován u 1 novorozence pro 20-25 tisíc.
První známky onemocnění se objevují během prvního roku života dítěte, ale úplný klinický obraz se může rozvinout až o 2 roky..
Nejtypičtější symptomy onemocnění jsou:
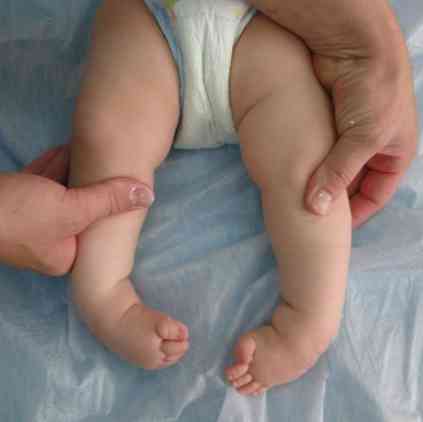 hrubé obrysy obličeje - velmi typické pro tuto patologii;
hrubé obrysy obličeje - velmi typické pro tuto patologii;- deformace lebky - stává se jako lodní kýl. Tento stav se nazývá scaracephalia;
- dýchání se provádí ústy, jelikož je zhoršování nosního dýchání způsobeno nárůstem adenoidů a intrauterinními vývojovými abnormalitami v lebce obličeje;
- deformace končetin a některých dalších fragmentů kostry;
- retardace růstu;
- zakřivení páteře (příznak "kočka zad" v sedě);
- zkrácení krku;
- atypické vysoké uspořádání lopatek;
- vyklenuté přední spodní žebra;
- zvýšení šířky kartáče - zatímco oni připomínají dráp s dráp;
- vývoj kontrakcí (snížení rozsahu pohybu) s postupným zatahováním různých kloubů do patologického procesu. Za prvé, jsou ovlivněny klouby na loktech a ramenou, pak se proces rozšiřuje na kolenní, kyčelní a kotníkové klouby.
 hrubé obrysy obličeje - velmi typické pro tuto patologii;
hrubé obrysy obličeje - velmi typické pro tuto patologii;Hurlerův syndrom se vyznačuje specifickou chůzí - na špičkách s ohnutými nohami. Takový rys vzniká kvůli omezení pohybu kvůli kontrakci..
Fyzikální vyšetření odhaluje následující:
- břicho je zvětšena, jeho přední stěna u těchto pacientů je oslabena, proto se mohou často vyvinout pupečníkové a inguinální kýly;
- játra a slezina rozšířené;
- U mužů je často detekována hydrocela - varlata;
- v některých případech jsou zjištěna porušení koordinace pohybů;
- odhalil nadměrný růst srstnatých vlasů.
Při instrumentálním vyšetření je určeno následující:
- na elektrokardiogramu - známky difúzního (rozšířeného) poškození srdečního svalu;
- poškození zraku a sluchu;
- oftalmoskopie odhaluje zákal a zvětšení rohovky.
Nejcharakterističtější jsou změny, které se vyskytují při rentgenovém vyšetření:
- RTG páteře - umožňuje identifikovat charakteristickou deformitu páteře. V tomto případě obratle mají tvar krychle, jejich okraje jsou zaoblené. Také výrazná kyfóza - zakřivení hřbetu vyčnívající vpřed;
- rentgenové vyšetření hrudníku - výrazné zesílení předních rebrových fragmentů se současným ztenčením zadních částí, zkrácení a deformace. Žebra současně získají zvláštní vzhled a podobají se lopatě nebo šavle. Zaznamenává se také pokles a posunutí hlavy humeru;
- radiografie pánve - odhaluje zúžení pánevního kroužku, acetabulární dutiny (ty, ve kterých je "základ" tvoření kyčelního kloubu), jsou zkosené a hlavy femur jsou sníženy;
- RTG trubičkových kostí - zeslabení kortikální (povrchní) vrstvy kostí a určitá expanze medulárního kanálu;
- RTG kostí na ruce - pomáhá detekovat nedostatečné rozvinutí distálních (nehtů) falangů prstů, zkracování a rozšíření středních a proximálních falangů, stejně jako metacarpálních kostí;
- RTG lebky - zřetelně nedostatečně vyvinuté kosti lebky obličeje, kraniostenóza (zúžení lebky v některých místech) a makrocefalie (rozšíření lebky).
U pacientů s diagnózou mukopolysacharidózy typu IH jsou identifikovány následující poruchy, které lze považovat za samostatné onemocnění:
- retinální pigmentová dystrofie - porušení jejího vývoje;
- glaukom - zvýšení nitroočního tlaku;
- přetížení v podzemí;
- paréza - narušení pohybu;
- paralýza - úplné narušení motorické aktivity v určitých oblastech muskuloskeletálního systému.
Časem se pacienti s diagnózou Hurlera vyvíjejí a nezvratně pokročily v mentální retardaci.
Mukopolysacharidóza typu I-S
Dalším názvem mukopolysacharidózy typu I-S je onemocnění onemocnění. Toto onemocnění bylo popsáno teprve v polovině 20. století, přestože pacienti s charakteristickými kontrakcemi prstů rukou, pohybovými poruchami ve velkých kloubech a specifickými rysy obličeje se setkali v praxi kliniků dříve. Symptomy mohou být kombinovány se známkami syndromu Hurler.
Tento typ mukopolysacharidózy se objevuje v pozdějším věku než u mukopolysacharidózy typu IH a jeho průběh je výhodnější..
Charakteristickým znakem mukopolysacharidózy typu I-S je to, že do 3 až 6 let věku vývoj dítěte odpovídá věkové normě. Dále se začínají objevovat následující znaky:
- flexi kontrasty prstů;
- omezení rozšíření ve velkých a středních kloubech - jmenovitě lak, rameno a zápěstí.
Porušení amplitudy pohybů na straně dolních končetin je méně výrazné než u jiných typů popsané patologie.
Kompletní klinický obraz tohoto typu mukopolysacharidózy je stanoven počátkem dospívání.
Fyziologická studie (vyšetření) uvádí následující:
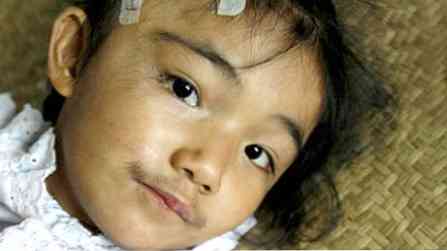 pacienti nejsou vysokí, ale mají silnou stavbu;
pacienti nejsou vysokí, ale mají silnou stavbu;- vlastnosti jsou spíše hrubé;
- kostní svalstvo dobře vyvinuté;
- hypertrichóza je zaznamenána (zvýšené srsti);
- kůže na prstech rukou (častěji) a nohy (méně často) je zhrubnutá a natažená;
- velikost jater a sleziny se zpravidla nezměnila.
 pacienti nejsou vysokí, ale mají silnou stavbu;
pacienti nejsou vysokí, ale mají silnou stavbu;Navíc jsou tito pacienti identifikováni porušení, které lze považovat za samostatnou nemoc:
- inguinální nebo pupeční hernie;
- syndrom karpálního tunelu - vzniká v důsledku výrazného stlačení mediálního nervu. To se projevuje atrofií (hypoplázie) tenárních svalů (vyvýšení v oblasti palce) a parestézie (porucha citlivosti) v oblasti 3, 4 a 5 prstů;
- aortální stenóza - zúžení aorty;
- aortální nedostatečnost;
- retinální pigmentová dystrofie;
- glaukom;
- zákal rohovky.
Inteligence pacientů s diagnostikovanou mukopolysacharidózou typu I-S zůstává normální, na rozdíl od pacientů s mukopolysacharidózou typu IH.
Z nejdůležitějších nástrojů je radiografická. Identifikuje změny, které jsou podobné změnám v mukopolysacharidóze typu IH, ale jsou méně výrazné..
Mukopolysacharidóza typu II
Tento typ mukopolysacharidózy se také nazývá Hunterova nemoc. Patologie nejčastěji postihuje chlapce ve věku 2-3 let.
Podle klinických projevů je onemocnění podobné mukopolysacharidóze typu IH - zatímco existují:
- scariocephaly;
- hrubé rysy;
- nízký tón hlasu;
- potíže s dechem - v důsledku deformace kostí obličejové kostry.
Při dalším postupu onemocnění se spojují následující příznaky:
- snížená inteligence;
- koordinace pohybů je přerušena;
- Nálada u takového pacienta je nestabilní - zaznamenává se takzvaná emoční labilita - od záchvatů smíchu až k pláči. Takové změny nálady se mohou kdykoli objevit..
S progresí mukopolysacharidózy typu II se objevuje a zvyšuje neoprávněná agresivita.
Na rozdíl od mukopolysacharidózy typu IH, porušení páteře ve formě kyfózy není charakteristické, příznak "kočky zpět" není pozorován.
Fyzikální vyšetření určuje následující:
- je zaznamenán mírný nárůst sleziny a jater;
- na pokožce zadní části jsou určeny specifické uzliny.
Když instrumentální vyšetření odhalilo:
- opacita rohovky, která postupuje;
- zvyšuje ztrátu sluchu.
Ze všech instrumentálních metod diagnózy je nejdůležitější radiografie různých fragmentů muskuloskeletálního systému - lebky, rudy, pánve a tak dále. Zároveň jsou radiologické znaky stejné jako u mukopolysacharidózy typu I-S.
Pacienti s takovou diagnózou jsou náchylní k vývoji onemocnění dýchacího systému:
- tracheitida;
- bronchitida;
- pneumonie.
Je třeba poznamenat, že mukopolysacharidóza typu II se může vyvíjet ve formě dvou možností:
- nepříznivé (možnost A);
- příznivé (možnost B).
 Nepříznivá varianta je zobrazena jasným klinickým obrazem a současně jsou zaznamenány podstatné poruchy inteligence. Během dospívání může dojít k úmrtí.
Nepříznivá varianta je zobrazena jasným klinickým obrazem a současně jsou zaznamenány podstatné poruchy inteligence. Během dospívání může dojít k úmrtí.
Symptomy s nepříznivým varianta, která nebyla vyjádřena, mohou být výše uvedenými porušeními doprovázena lehkou mentální retardací. Tito pacienti zpravidla žijí na 30 let, v některých případech - a více.
Mukopolysacharidóza typu III
Mucopolysacharidóza typu III byla popsána americkým pediatrem Sanfillipem, takže patologie dostala jiný název - onemocnění Sanfillipo.
Nemoc je diagnostikován u 1 dítěte na 100-200 tisíc novorozenců..
Děti s mukopolysacharidózou typu III se v prvních letech svého života vyvíjejí podle věkových norem a mohou být pozorovány některé patologické příznaky - obzvláště potíže s polykáním a nepříjemná chůze..
Od 3-5 let začíná dítě zaostávat za svými vrstevníky ve fyzickém vývoji, zároveň se stává apatická, lhostejná vůči lidem a událostem.. Typické znaky jsou:
- porucha řeči;
- drsnost prvků;
- pravidelná močová inkontinence (nejen noční) a výkaly.
Jak dítě roste, mentální retardace se zvětšuje a nakonec se stává hlavním příznakem tohoto typu mukopolysacharidózy..
Fyzikální vyšetření ukázalo:
- zanedbatelné zvětšení jater a sleziny;
- jednotné zvýšení rozložení vlasů;
- kontraktura;
- zpomalení růstu.
 Porušení organu vidění a kardiovaskulárního systému, které se často vyskytují v jiných formách popsané patologie, pro tento typ mukopolysacharidózy nejsou typické.
Porušení organu vidění a kardiovaskulárního systému, které se často vyskytují v jiných formách popsané patologie, pro tento typ mukopolysacharidózy nejsou typické.
Výsledky rentgenového vyšetření s popsanou patologií nebo bez patologických změn, nebo podobné výsledky, které byly detekovány s mukopolysacharidózou typu I-S, ale méně výrazné.
Prognóza této choroby je méně příznivá než u jiných typů mukopolysacharlózy - tito pacienti zemřou ve věku 10 až 20 let, infekční komplikace se stávají příčinou smrti..
Mukopolysacharidóza typu IV
Mucopolysacharidóza typu IV se také nazývá Morkio choroba (pojmenovaná podle uruguayského pediatra, který ji popsal). Patologie je častější než jiné typy mukopolysacharidózy - jmenovitě u 1 dítěte na 40 tisíc novorozenců.
Děti s touto chorobou do 1-3 let se vyvíjejí bez vlastností. Poté se objeví následující případy porušení:
- dítě výrazně zaostává za svými vrstevníky;
- v procesu vývoje zaznamenal zkrácení krku a trupu;
- rozvíjí se různé kontrakty;
- kůže se ztuhne;
- svalová síla klesá;
- sluch se zhoršuje;
- obličejové rysy se stávají drsnými;
- se vyskytují různé hrudní deformity.
Také u mukopolysacharidózy typu IV existují porušení, které lze považovat za samostatné patologie:
- valgusová deformita končetin - zakřivení směrem ven;
- skolióza (zakřivení páteře v laterální rovině) nebo kyfóza;
- inguinální kýla;
- pupeční hernie;
- dystrofie rohovky.
Intelektu s touto formou mukopolysacharidózy zachránil.
Diagnóza popsané patologie je potvrzena rentgenem, je v tomto případě nejvíce informativním. Jedná se o následující typy:
- Rentgenové vyšetření páteře - obrazy ukazují zakřivení páteře ve formě kyfózy a skoliózy, stejně jako rozšíření a zploštění těl obratlů;
- radiografie pánve - odhalilo několik deformací pánevního kostního kruhu, nerovnoměrnost jeho obrysů;
- Rentgenové vyšetření končetin - zploštění hlavy femuru, výrazné zkrácení kosti předloktí a deformita kostí nohou.
Pacienti s mukopolysacharidózou typu IV žijí zpravidla méně než 20 let. Smrtelný výsledek se objevuje na pozadí mnoha souvisejících onemocnění, které jsou v konečném důsledku komplikované kardiopulmonální nedostatečností..
Mukopolysacharidóza typu VI
Dalším názvem pro mukopolysacharidózu typu VI je Maroto-Lamyova choroba (po jménech dvou francouzských lékařů, kteří popsali tuto patologii)..
Klinické příznaky se většinou "ozývají" příznaky jiných typů mukopolysacharidózy, ale nejvýraznější jsou:
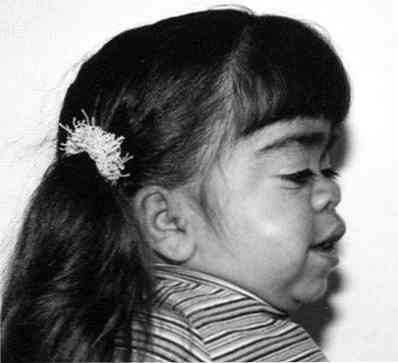 významné zvlnění tváře;
významné zvlnění tváře;- výrazné zpoždění růstu;
- zkrácení krku - někdy se zdá, že pacientova hlava "sedí" téměř na ramínku;
- deformita hrudníku.
 významné zvlnění tváře;
významné zvlnění tváře;Tito pacienti mají velmi často nachlazení - tento příznak umožňuje podezření na tuto konkrétní, a ne jinou formu mukopolysacharidózy..
Fyzikální vyšetření odhaluje následující:
- játra a slezina jsou často zvětšeny;
- může být diagnostikována inguinální kýla.
Inteligentně se dítě vyvíjí bez porušení - má schopnost přemýšlet a řešit intelektuální problémy..
Existují dvě varianty tohoto typu mukopolysacharidózy:
- klasické;
- mukopolysacharidóza s malými klinickými projevy.
Pro potvrzení diagnózy pomůže rentgenová vyšetření - snímky jsou určeny:
- deformace obratlů - stávají se kvádrovými nebo klínovitými;
- trojúhelníková deformace pánevního kroužku;
- hypoplázie (nedostatečné rozvinutí) a deformace hlavy femuru;
- zkrácení kosti fibula.
Mukopolysacharidózy typu VII a VIII
 Mucopolysacharidóza těchto typů je méně častá než jiné typy popsané patologie..
Mucopolysacharidóza těchto typů je méně častá než jiné typy popsané patologie..
Mucopolysacharidóza typu VII klinicky se vyskytuje jako onemocnění typu III, rozdíl je mezi nimi pouze ve výsledcích biochemických studií.
Mucopolysacharidóza typu VIII je symptomatická a podobná mukopolysacharidóze typu IV. Existuje však rozdíl v projevech - je to narušení intelektuálního vývoje, které se nepozoruje u mukopolysacharidózy typu IV.
Diagnostika
Při diagnostice mukopolysacharidózy jsou založeny na stížnostech, anamnestických datech, výsledcích dalších vyšetřovacích metod - fyzikálních, instrumentálních, laboratorních. Lze předpokládat přítomnost mukopolysacharidózy charakteristickými deformacemi kostry, které "chytají oko", ale je možné určit typ popsané nemoci pouze pomocí dodatečných vyšetřovacích metod. Hlavní metody jsou:
- radiografie;
- detekce glykosaminoglykanů v moči;
- stanovení enzymatické aktivity v buněčných kulturách.
Pokud klinické příznaky poruch vnitřních orgánů zahrnují metody vyšetření:
- elektrokardiografie;
- ultrazvuk jater a sleziny;
- elektroencefalografie;
- biochemický krevní test
a mnoho dalších.
Diferenciální diagnostika
Diferenciální (specifická) diagnóza se provádí mezi různými typy mukopolysacharidózy.
Komplikace
Nejčastějšími komplikacemi mukopolysacharidózy jsou:
- kontraktura;
- srdeční selhání;
- plicní nedostatečnost.
Léčba
Terapie na mukopolysacharidózu, která může být použita k odstranění onemocnění, a nikoliv pouze jejích projevů, nebyla vyvinuta. Tito pacienti dostávají symptomatickou léčbu - mohou to být:
- konzervativní - krevní transfúze, hormonální terapie, terapie vitaminem a podobně;
- operativní - endoprotéza hlavy femuru, oprava kýly, chirurgická intervence k odstranění kontrakcí a správné kosterní deformity a další.
Důležité jsou:
- léčba infekcí dýchacích cest;
- korekce zrakových a sluchových poruch.
Prevence
Vzhledem k tomu, že patologie vznikají v důsledku genetických poruch, neexistují žádné specifické metody prevence.. Ale riziko genových poruch, které mohou vést k rozvoji různých typů mukopolysacharidózy, může být sníženo, pokud je ženě poskytnuto normální těhotenství. Toto je:
- odmítnutí špatných návyků;
- dodržování práce, odpočinku, spánku;
- optimistická nálada.
Předpověď

Prognóza mukopolysacharidózy je obecně nepříznivá, což se týká všech typů popsaných onemocnění. V průběhu života a vývoji v tkáních se postupně akumulují metabolické produkty, které vznikají kvůli nedostatku tkáňových enzymů. To vede k patologickým změnám ze strany téměř všech orgánů a systémů. Pokud dojde k patologii, smrt může nastat nejen kvůli kardiopulmonálním, ale i polysystémovým selháním.
Konzervativní terapie hraje roli podpůrné - použití jakéhokoli léku vede k dočasnému zlepšení, ale není schopné kompenzovat nedostatek tkáňových enzymů.
Kovtonyuk Oksana Vladimirovna, lékařský komentátor, chirurg, konzultant lékař